Background
Facioscapulohumeral dystrophy (FSHD) is one of the most common types of muscular dystrophy. It has distinct regional involvement and progression. FSHD is an autosomal dominant disorder in as many as 90% of affected patients. Landouzy and Dejerine first described FSHD in 1884. Tyler and Stephens described an extensive family from Utah in which 6 generations were affected. Walton and Nattrass established FSHD as a distinct muscular dystrophy with specific diagnostic criteria.
Pathophysiology
It is an autosomal dominant disease in 70-90% of patients and is sporadic in the rest. One of the FSHD genes has been localized to chromosome band 4q35, but the gene or genes that are affected in FSHD are still unknown. Patients with FSHD have a shorter Eco RI digestion fragment detected by the chromosome-4qter DNA marker p13E-11. About 2% of FSHD patients are not linked to the locus at 4q35.
The probe p13E-11 identifies 2 polymorphic loci at 4q35 and 10q26. The Eco R1 fragment of 4q is composed of repetitive DNA sequences that are 3.3-kilobase (kb)Kpn I tandem repeats identified as D4Z4. In control subjects, the D4Z4 repeat consists of 11-100 KpnI units, each 3.3 kb, whereas in FSHD this is shortened; the shortened Eco RI fragment in FSHD is 1-10 units. Diagnostic difficulties arise as these fragments also may come from chromosome 10, as already described. 4-Type units are resistant to Bln I and 10-type units are resistant to Xap I. The combined use of EcoRI, BlnI, and XapI in pulsed-field gel electrophoresis–based DNA separation techniques allows detection of 4q fragments.
- FSHD is caused by a contraction mutation of D4Z4 macrosatellite repeats in the subtelomeric region of the 4qA161 haplotype of chromosome 4 in 95% of patients.
- Those without FSHD have approximately 11-100 D4Z4 units, whereas patients with FSHD have 1-10 D4Z4 units.
- At least 1 copy of D4Z4 is required to develop FSHD.
- Mosaic males are mostly affected, where as mosaic females with an equal complement of affected cells are more often asymptomatic carriers.
- A bi-allelic variation of chromosome 4qter is known, designated as 4qA and 4qB. FSHD alleles are exclusively of the 4qter type (4qA161).
- Although the genetic lesion is well described in FSHD, the causal gene and the protein products are not known.
- Additional testing may be needed in patients without D4Z4 contraction for a deletion encompassing the region.
- Patients with FSHD and no contraction of D4Z4 repeats may show loss of DNA methylation and heterochromatin markers of D4Z4 repeat.
- The most extensively studies candidate genes for FSHD on 4q35 are ANT1, PDLIM3, FRG1, TUBB4q, FRG2, and DUX4.
Disease mechanisms
The actual genetic defect in FSHD is unknown. Possible disease mechanisms include the following:
- Position variegation effect on a proximal candidate gene or genes
- Direct and indirect evidence points to epigenetic modifications in the DNA. A local deficit of a repressor complex due to the contraction of D4Z4 may cause inappropriate expression of genes. This may account for upregulation of FRG2, FRG1, and ANT1 in FSHD muscle.
- The most common modification of mammalian DNA is cytosine methylation that is necessary for many regulatory processes. D4Z4 was found to be hypomethylated in FSHD.
- Myoblasts from patients with FSHD also demonstrate increased susceptibility to oxidative stress.
- Misexpression of FRG1 (FSHD region gene 1) may lead to the development of FSHD. Knockdown of FRG1 in Xenopus led to the decreased angiogenesis and reduced expression of DAB2 (angiogenic regulator). Of patients with FSHD, 50-75% exhibit retinal vasculopathy and increased expression of vascular or endothelial-related FRG1 transcripts in the muscle. Thus FRG1 may be at least crucial for angiogenesis.
- Deletion of D4Z4 macrosatellites results in aberrant gene expression. DUX4 transcript from the last D4Z4 (most telomeric) unit generates small si/miRNA-sized fragments; uncapped, polyadenylated 3-prime fragments encoding C-terminal portion of DUX4; capped and polyadenylated mRNAs containing the double-homeobox domain of DUX4, but splice-out the C-terminal polypeptide. C-terminal polypeptide produced by transfection studies inhibits myogenesis.[1]
- DUX4 is a retrogene contained within D4Z4 repeats and is normally epigenetically silenced in somatic cells. D4Z4 contraction leads to loss of DNA methylation and heterochromatin markers in D4Z4 region, resulting in relaxation of chromatin structure and release DUX4 repression.[2]
Epidemiology
Frequency
United States
FSHD is the third most common muscular dystrophy. Estimated prevalence of FSHD is 1 case in 20,000 persons.
Mortality/Morbidity
Most of the patients have normal life expectancy.
Sex
Frequency is higher in males; however, asymptomatic cases are more common in females.
Age
- The usual presentation is between the first and third decades. Ninety-five percent of patients show clinical features before age 20 years. As many as one third of patients are asymptomatic.
- Infantile onset has been described, but is rarePRESENTATION
Physical
.- Initial weakness is seen in facial muscles, starting in the orbicularis oculi, orbicularis oris, and zygomaticus.
- Patients may have difficulty with labial sounds, whistling, or drinking through a straw.
- Weakness may be asymmetric.
- Extraocular and pharyngeal muscles are spared.
- Shoulder weakness is the presenting symptom in more than 82% of patients with symptoms.
- Scapular fixation is weak from the onset. Winging of the scapula is the most characteristic sign. The scapula is placed more laterally than normal. It moves upwards in shoulder abduction.
- The deltoid muscle usually is spared, and shoulder abduction weakness is predominantly due to weak scapula fixation.
- If the scapula is stabilized manually against the chest wall, the patient may experience improved movement. Upward slope of the anterior axillary fold results from weakness of the pectoralis major.
- Truncal weakness is early. Lower abdominal muscles are weaker than upper abdominal muscles, resulting in the Beevor sign, a physical finding very specific for FSHD. The Beevor sign is the upward movement of the umbilicus toward the head when flexing the neck.
- Weakness of foot dorsiflexion follows shoulder weakness.
- Tibialis anterior muscle weakness is highly characteristic, whereas posterior muscles of the leg are spared.
- In a few patients, a foot-drop gait is the presenting complaint. In more than 50% of patients, the pelvic girdle muscles are never involved.
- Atypical phenotypes in patients with FSHD
- Scapulohumeral dystrophy (SHD) or facial-sparing SHD with or without myalgia
- FSHD with chronic progressive external ophthalmoplegia (CPEO)
- Limb-girdle muscular dystrophy syndrome
- Distal myopathy
- Asymmetric brachial weakness
- Extramuscular manifestations are as follows:
- High-frequency hearing loss in almost 75%
- Retinal telangiectasias in about 60%
- Atrial arrhythmias in 5%
- Restrictive respiratory disease in 1%
- Mental retardation
- Seizures
- Sleep-disordered breathing (SDB) is very common in FSHD. Obstructive sleep apnea, REM-related oxygen desaturation, or mixed pattern were observed in 39% of FSHD patients. SDB is not related to severity of the disease.[3
Differential Diagnoses
- Amyotrophic Lateral Sclerosis
- Chronic Inflammatory Demyelinating Polyradiculoneuropathy
- Congenital Muscular Dystrophy
- Congenital Myopathies
- Dermatomyositis/Polymyositis
- Diabetic Neuropathy
- Emery-Dreifuss Muscular Dystrophy
- Endocrine Myopathies
- Inclusion Body Myositis
- Inherited Metabolic Disorders
- Limb-Girdle Muscular Dystrophy
Laboratory Studies
Serum creatine kinase levels are elevated.Imaging Studies
Imaging studies show a selective destructive process involving the anterior compartment muscles of the leg. Hypertrophy of the psoas muscles also is observed occasionally.Other Tests
- FSHD gene testing
- Electrodiagnostic studies; these may reveal myopathic potentials. Focal neuropathies and occasionally a brachial plexopathy may be seen as a result of stretch injury.
Procedures
Muscle biopsy: If results of genetic testing for FSHD are negative, a muscle biopsy is strongly recommended to rule out other conditions that mimic FSHD.Histologic Findings
- Muscle
- Multifocal distribution
- Fiber size variation with central nuclei
- Endomysial connective tissue proliferation
- Regenerating, moth-eaten fibers and muscle fibers undergoing phagocytosis
- Isolated angular atrophic fibers
- Rimmed vacuoles
- Focal inflammation in perivascular and perimysial distributions
- Muscle fibers expressing membrane attack complex
- Muscle fibers expressing class 1 major histocompatibility antigen
Medical Care
- No definitive therapy is available for FSHD.
- Custom-made ankle-foot orthosis (AFO) may help patients with prominent foot drop. Sometimes AFO may worsen the gait in the presence of knee extensor weakness and these patients may benefit from floor reaction AFO (FRAFO) or newer knee-ankle-foot-orthosis (KAFO).[4]
- Corticosteroids failed to improve muscle strength or muscle mass.
- A pilot trial of sustained-release albuterol taken PO (16 mg/d) for 3 months increased lean body mass. A modest 12% increase in muscle strength was noted.
- A double-blind placebo-controlled trail randomizing the patients to placebo, 8 mg albuterol twice daily, or 16 mg albuterol twice daily showed no improvement in global strength. However, albuterol improved grip strength and muscle mass. Basing on the information available, albuterol cannot be recommended.
- In a randomized, double-blinded, cross-over trial in a mixed population of dystrophies (12 with FSHD), a creatine monohydrate value of 10 g/d demonstrated a slight improvement in overall strength.
- Payan et al studied the effect of salbutamol on muscle strength in patients with genetically confirmed FSHD. Ambulatory patients received either salbutamol (n=56) or placebo (n=56) for 6 months. No significant change in muscle strength was shown with salbutamol compared with placebo. Results from this study do not support the routine use of salbutamol for FSHD.[5]
- Creatine monohydrate, folic acid and methionine supplementation and myostatin inhibition (MYO-29) have been tried with no benefit.
- Aerobic training may improve exercise performance.[6] Twelve weeks of low-intensity aerobic exercises (on a cycle ergometer at a heart rate corresponding to a work intensity of 65% of VO2 max for 35-min weekly sessions and increased to 5-times weekly in 4 wk) improved maximal oxygen uptake and work load with no signs of muscle damage.
Surgical Care
Scapulothoracic arthrodesis may be attempted in select patients with preserved deltoid function. An improved functional range of abduction can be achieved if the scapula is fixed in 15-20° of rotation. In a series by Bunch and Siegel, 11 of 12 patients improved with this procedure.[7]Demirhan, using multifilament cable for scapulothoracic arthrodesis, provided satisfactory function (Level IV evidence) in 13 patients.[8]Medication Summary
The goals of pharmacotherapy are to reduce morbidity and prevent complications.Beta2-adrenergic agonists
Class Summary
By activating cyclic AMP, this agent stimulates the ATPase pump, thereby activating the beta-adrenergic pathway. Albuterol reportedly improves muscle strength in FSHD patients through its nonspecific anabolic properties.Albuterol (Ventolin, Proventil)
Relaxes bronchial smooth muscle by action on beta2-receptors with little effect on cardiac muscle contractility.Complications
- Coats syndrome: This syndrome, a retinal vasculopathy with telangiectasia, exudation, and retinal detachment, is seen in 49-75% of affected individuals. If detected early, retinal photocoagulation may prevent serious consequences.
- Hearing loss: Sensorineural deafness is observed in 64% of patients; it may be unilateral.
- Mental impairment and epilepsy: These are seen in the early onset group. Mental retardation is observed in about 40% of patients with early onset 4q35-FSHD. Epilepsy also is observed often in this subset of patients.
- Labile hypertension
- Cardiac complications: Atrial arrest, bundle branch block, and dilated cardiomyopathy have been reported.
Prognosis
- Size of deletion affects disease severity and thus prognosis. Ricci studied 122 Italian families affected by FSHD and 230 healthy control subjects. An Eco RI fragment shorter than 30 kb that was resistant to Bln I restriction was found in 114 of 122 families (93%) with FSHD. Fifteen percent of the control group showedEco RI fragments smaller than 30 kb that were Bln I sensitive, suggesting that these were 10 qter alleles. Prognosis varied with the length of the fragment size and the remaining Kpn I units.[9] The probabilities of developing the severe form of the disease were as follows:
- 100% with very short segment length of 10-13 kb (1-2 Knp I repeats left)
- 54% in patients with fragment length of 16-20 kb (3-4 Knp I repeats left)
- 19% in patients with fragment length greater than 21 kb (more than 4 Knp I repeats left)
- Age of onset is variable. The disease tends to progress from the face downwards. Asymmetry and selective muscle group involvement distinguish FSHD from other muscular dystrophies. Many authors describe stepwise deterioration with prolonged periods of apparent arrest. Extraocular muscles, bulbar muscles, deltoids, and respiratory muscles usually are spared. Ventilatory impairment is seen in fewer than 10% of patients.
- Approximately 20% of patients may require wheelchair assistance.
- Life expectancy is normal in most patients.
 27-27year-old female with faciohumeroscapular muscular dystrophy. Marked non-structural hyperlordosis. Upright position achieved only by forward pelvic tilt
27-27year-old female with faciohumeroscapular muscular dystrophy. Marked non-structural hyperlordosis. Upright position achieved only by forward pelvic tilt
SCAPULAR WINGING
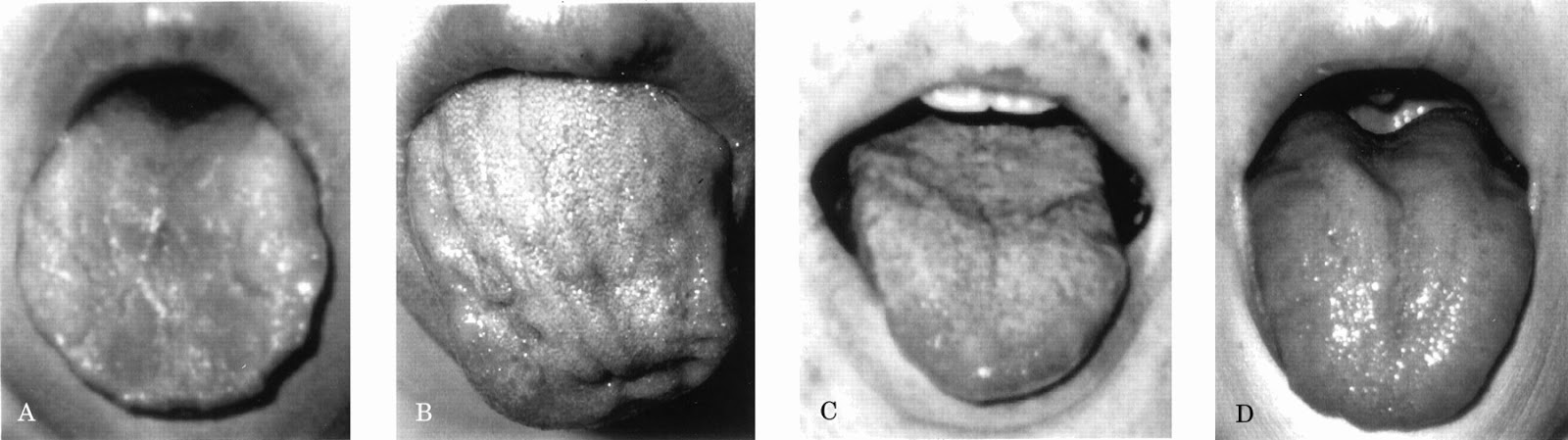 Tongue atrophy in facioscapulohumeral muscular dystrophy
Tongue atrophy in facioscapulohumeral muscular dystrophy
Asymmetric triangular shoulders
 Retinal Vasculopathy associated with Facioscapulohumeral Muscular Dystrophyhttp://www.youtube.com/watch?v=QlF25CBNR3Y
Retinal Vasculopathy associated with Facioscapulohumeral Muscular Dystrophyhttp://www.youtube.com/watch?v=QlF25CBNR3Y
Internet Links to other FSH sites
its really informative .. jzak allah khair
ردحذفits really informative .. jzak allah khair
ردحذف